Malattie Rare e Farmaci Orfani: il settore rimane uno dei più interessanti per le aziende, e dovrebbe essere rinnovato dalla revisione della legislazione farmaceutica europea ancora in corso
I bisogni medici insoddisfatti hanno rappresentato uno degli ambiti di ricerca e sviluppo di nuovi medicinali più attrattivi per le aziende farmaceutiche nel corso dell’ultimo trentennio.
In questo contesto, un posto di tutto rilievo è quello dei farmaci destinati al trattamento delle cosiddette “malattie rare”, ovvero quelle malattie che colpiscono – secondo la definizione che ne dà l’Unione Europea – meno di cinque persone ogni 10 mila.
L’esiguità delle popolazioni di pazienti ha storicamente reso poco interessante sul piano economico lo sviluppo di medicinali dedicati alle malattie rare, una vasta famiglia di prodotti che viene nel suo complesso indicata col termine “farmaci orfani”.
Se è vero che a ogni malattia rara corrisponde una assai piccola popolazione di pazienti, lo scenario potrebbe cambiare se si va a osservare il quadro complessivo: secondo i dati dell’Agenzia europea dei medicinali (EMA), infatti, alle oltre 6 mila malattie rare conosciute corrisponderebbe nell’Unione Europea un volume complessivo di potenziali pazienti pari a circa 26 milioni di persone (un paziente ogni 17 abitanti).
Un dato che deve venire considerato anche alla luce del fatto che questo tipo di prodotti riesce spesso a spuntare prezzi molto elevati in fase di contrattazione tra le aziende produttrici e le autorità competenti a livello nazionale, oltre che dal poter godere di numerosi incentivi per il loro sviluppo.
Designazione è diverso da autorizzazione
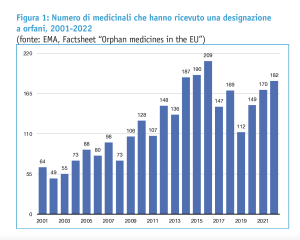
Secondo gli ultimi dati EMA, il numero di medicinali che hanno ricevuto la designazione a farmaco orfano nel periodo 2000-2022 è di oltre 2.730 prodotti.
Sono però solo poco più di 230 i medicinali orfani autorizzati per l’immissione in commercio sul mercato dell’Unione europea nello stesso periodo.
Non è detto, quindi, che un medicinale che sia stato designato come farmaco orfano da parte del Committee for Orphan Medicinal Products (COMP) di EMA raggiunga anche lo stadio di deposito della domanda di autorizzazione AIC e passi la conseguente valutazione da parte del Comitato EMA per i medicinali a uso umano (CHMP).
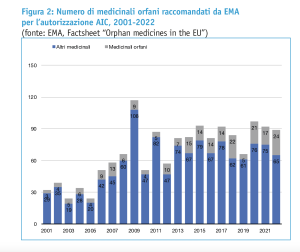
A fronte dei 182 prodotti che hanno ricevuto la designazione a farmaco orfano nel corso del 2022, ad esempio, solo 24 medicinali sono stati raccomandati dal CHMP per l’autorizzazione all’immissione in commercio da parte della Commissione europea.
Tutti i medicinali che hanno ricevuto una designazione a farmaco orfano sono elencati, inclusa l’indicazione, nell’apposita sezione del Registro dell’Unione dei prodotti medicinali (Community Register of orphan medicinal products, accessibile online al link https://ec.europa.eu/health/documents/community-register/html/reg_od_act.htm?sort=n)
Oltre che il criterio numerico sulla popolazione di pazienti sopra menzionato, la designazione a farmaco orfano da parte del comitato COMP prende anche in considerazione la gravità della malattia, la mancanza di ritorni economici per le aziende sviluppatrici e l’assenza di metodi soddisfacenti per la diagnosi, prevenzione o trattamento della patologia (o, ove esistenti, l’apporto di un beneficio addizionale significativo).
Non rientrano, invece, nella definizione di farmaci orfani i prodotti di medicina personalizzata basati sull’utilizzo di biomarcatori per l’identificazione delle sottopopolazioni di pazienti su cui un certo medicinale è atteso essere efficace.
La presentazione delle domande di designazione a farmaco orfano rimane un processo tipico delle grandi aziende farmaceutiche: i dati EMA (“Orphan Medicinal Product Designation. Overview 2000-2022”) indicano per il 2022 un contributo al fenomeno da parte delle piccole e medie aziende pari al 26% del totale. La grande maggioranza dei prodotti in sviluppo designati come farmaci orfani, inoltre, sono prodotti di stampo più tradizionale (88%). Solo una piccola quota (12%) appartiene alla categoria delle terapie avanzate.
I due terzi circa dei prodotti, infine, negli ultimi anni hanno basato la designazione a farmaco orfano sul criterio del beneficio significativo (66% nel 2022).
Nello stesso anno, le nuove condizioni hanno rappresentato il 16% del totale delle designazioni.
Sempre con riferimento al 2022, le categorie terapeutiche più rappresentate sono state riferite ai prodotti per il trattamento di malattie congenite, familiari o genetiche (62), malattie del sistema nervoso (29) e malattie del sangue e del sistema linfatico (22).
Le domande di autorizzazione attive riferite a medicinali orfani seguono un andamento simile per le prime due posizioni (47 per malattie congenite e familiari, 23 per malattie del sangue e linfatiche), mentre al terzo posto si collocano i medicinali contro neoplasie benigne e maligne (22), seguiti da quelli per le infezioni (11).
La maggior parte dei prodotti designati come orfani sono destinati a malattie che colpiscono sia gli adulti che le popolazioni pediatriche; solo il 4% dei prodotti sono destinati in modo mirato unicamente a queste ultime.
Il processo di sviluppo
La designazione a farmaco orfano è un evento che si colloca all’inizio del percorso di sviluppo di un nuovo medicinale. Ciò implica che questi prodotti non possono normalmente essere impiegati sui pazienti (serve l’autorizzazione AIC), in quanto seppur promettenti potrebbero ancora mancare di sufficienti evidenze di efficacia.
Le Question and Answers pubblicate da EMA nel 2018 (EMA/551338/2017) specificano che è possibile in casi eccezionali un utilizzo dei farmaci designati orfani in base a programmi per uso compassionevole, ove non esistano altre possibilità per il paziente, ovvero potrebbero venire avviate sperimentazioni cliniche a cui potrebbe essere possibile partecipare.
La designazione a farmaco orfano, inoltre, non ha effetti sul tempo necessario a completare lo sviluppo.
Una volta ricevuta l’autorizzazione all’immissione in commercio, inoltre, il comitato COMP di EMA è chiamato a confermare o annullare la designazione a farmaco orfano sulla base delle nuove evidenze emerse nel corso dello sviluppo stesso.
Le designazioni ad opera di autorità regolatorie di altre aree geografiche, come l’americana FDA, non sono valide nell’Unione Europea, in quanto basate su criteri di valutazione (tra cui la soglia di prevalenza della malattia) diversi da quelli adottati in Europa. Gli accordi di confidenzialità in essere tra EMA e FDA permettono, invece, di condividere informazioni sui medicinali orfani. Le autorità regolatorie sulle due sponde dell’Atlantico, inoltre, hanno sviluppato procedure comuni per presentare le domande di designazione e i report annuali sullo stato dello sviluppo dei farmaci designati orfani.
Un elemento significativo dello sviluppo di un farmaco orfano è rappresentato dalla dimostrazione del “beneficio significativo”, che si differenzia dalla dimostrazione del rapporto beneficio-rischio richiesta in generale per tutti i medicinali. I farmaci orfani devono apportare benefici aggiuntivi rispetto ai trattamenti già disponibili, ad esempio portare a esiti migliori o consentire modalità di somministrazione più facili o agevoli. Il rapporto beneficio-rischio, invece, si basa sulla dimostrazione che gli esiti di salute ottenibili con la somministrazione del medicinale superano gli effetti avversi ad esso correlati.
Una volta ottenuta l’autorizzazione all’immissione in commercio con procedura centralizzata gestita da EMA, l’effettiva disponibilità del medicinale orfano nei diversi paesi membri dell’UE dipende dai passaggi di negoziazione di prezzo e rimborso che sono in carico alle singole autorità competenti a livello nazionale. Le differenze dei criteri di health technology assessment, utilizzati nei diversi paesi per definire i requisiti e le priorità dei rispettivi sistemi sanitari, sottolineano le Q&A di EMA, spiegano il fatto che a volte un certo farmaco orfano sia disponibile solo in alcuni paesi e non in altri. A questo livello, possono entrare in gioco anche comparazioni con altre tecnologie sanitarie o valutazioni value-for-money rispetto all’impatto atteso per il sistema sanitario del paese.
Gli attuali incentivi
Secondo la legislazione europea sui farmaci orfani attualmente in vigore (Regolamento europeo (EC) 141/2000), una volta ottenuta l’autorizzazione all’immissione in commercio e confermata la designazione, questi prodotti possono godere di un periodo di esclusività di mercato pari a dieci anni, al fine di favorire i ritorni sugli investimenti delle aziende che hanno affrontato lo sviluppo. Altri due anni di estensione dell’esclusività di mercato conferita dai certificati di protezione complementare (SPC) sono concessi ai medicinali pediatrici autorizzati sulla base degli studi definiti all’interno di un Piano di indagine pediatrica (PIP).
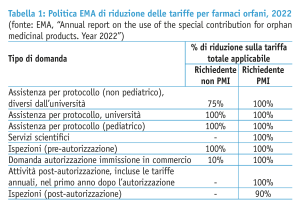
Questo è solo l’ultimo di una serie di incentivi pensati per sostenere le aziende lungo l’intero percorso di sviluppo. La designazione a farmaco orfano è un passaggio essenziale che permette di accedere, tra gli altri, ad attività di scientific advice e assistenza alla messa a punto dei protocolli sperimentali da parte di EMA pensate in modo mirato per questa tipologia di prodotti, oltre che a facilitazioni sulle tariffe regolatorie (in particolare per le micro, piccole e medie imprese e per gli sponsor universitari).
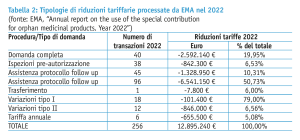
Più in particolare, ogni anno l’Unione europea destina un certo budget (il “contributo speciale”) a favore di EMA per coprire le riduzioni delle tariffe per i medicinali orfani (tabella 1). Gli ultimi dati relativi al 2022 indicano uno stanziamento totale di poco meno di 12,9 milioni di euro; il 61% delle tariffe hanno coperto attività di supporto allo sviluppo da parte dell’Agenzia, il 26% attività finalizzate all’approvazione regolatoria, incluse le ispezioni pre-autorizzazione (dati EMA, tabella 2).
Il percorso di sviluppo di ogni farmaco che ha ricevuto la designazione a orfano, infine, è monitorato dal comitato COMP di EMA per mezzo di un rapporto annuale che gli sponsor devono presentare all’Agenzia.
Gli incentivi previsti dalla revisione della legislazione farmaceutica
La proposta di regolamento pubblicata dalla Commissione il 26 aprile 2023 (COM(2023) 193 final), che è parte della più ampia manovra di revisione della legislazione farmaceutica europea, ridefinisce ad ampio raggio lo schema per la concessione degli incentivi per lo sviluppo di nuovi medicinali, compresi quelli destinati a malattie rare e farmaci orfani. In seguito alla conclusione del processo legislativo in corso, l’entrata in vigore del nuovo regolamento dovrebbe portare a una semplificazione normativa, con abrogazione dell’attuale regolamento sui farmaci orfani e di quello generale sui medicinali a uso umano ((EC) 726/2004), oltre che all’incorporazione di parti significative del regolamento sui farmaci pediatrici ((EC) No 1901/2006).
La bozza di regolamento si focalizza, in modo particolare sulle aree di cosiddetto “elevato bisogno medico insoddisfatto” (HUMN), caratterizzate da rischi e incertezze maggiori a livello delle attività di R&D. I criteri proposti per l’identificazione dei prodotti mirati ad esercitare i loro effetti terapeutici all’interno di tali aree prevedono l’assenza di medicinali già autorizzati per una certa patologia o l’apporto di eccezionali progressi terapeutici. Il nuovo farmaco dovrebbe, inoltre, apportare una significativa riduzione della morbilità o mortalità associate alla malattia.
Per quanto riguarda i farmaci orfani, i nuovi criteri di modulazione degli incentivi prevedono che essi possano godere di un periodo di esclusività di dieci anni nel caso si rivolgano a “elevati bisogni medici insoddisfatti”. Tale periodo si riduce a cinque anni nel caso di medicinali orfani di uso già ben consolidato, ed è in generale di nove anni per tutti gli altri farmaci orfani.
A ciò si potrebbe aggiungere un ulteriore anno di esclusiva qualora il medicinale venga commercializzato in tutti i paesi UE. Anche le prime due nuove indicazioni di medicinali orfani già autorizzati dovrebbero poter godere di un ulteriore anno di esclusività (che si estenderebbe all’intero prodotto, purché l’approvazione sia concessa almeno due anni prima dalla scadenza del periodo di esclusiva). In totale, quindi, i farmaci orfani dovrebbero poter godere, alla luce della nuova normativa, di un massimo di dodici anni di protezione del mercato.
Significativa, da questo punto di vista, risulterebbe anche l’introduzione di un approccio complessivo alle autorizzazioni AIC per una stessa sostanza attiva: qualora, infatti, il titolare AIC possieda più di una autorizzazione per la stessa sostanza, la bozza di regolamento prevede che venga conteggiato un unico periodo di esclusività a partire dalla data di concessione della prima autorizzazione all’immissione in commercio come farmaco orfano. È anche prevista la possibilità per i generici e biosimilari di ottenere un’autorizzazione AIC a partire da due anni di esclusività residua, così da favorire il tempestivo accesso al mercato al momento della scadenza finale.
Altre misure allo studio per i farmaci orfani, in particolare quelli per il trattamento di bisogni HUMN, prevedono l’ulteriore rafforzamento del supporto regolatorio e scientifico da parte di EMA e procedure di valutazione accelerata sul modello di quanto avvenuto con lo schema Prime e nel periodo Covid.
La prevista riorganizzazione dei comitati di EMA dovrebbe portare con sé anche il passaggio dalla Commissione europea alla stessa Agenzia europea dei medicinali per quanto riguarda la decisione finale circa la designazione a farmaco orfano. I medicinali orfani per la diagnosi, prevenzione o trattamento di malattie gravemente debilitanti o a pericolo di vita o da utilizzarsi in caso di situazioni di emergenza sanitaria dovrebbero rientrare tra le categorie per le quali sarà possibile concedere autorizzazioni AIC condizionate sulla base di dati ancora incompleti.
EMA potrà anche raccomandare alla Commissione l’adozione di criteri di designazione a farmaco orfano particolari per specifiche malattie, qualora quelli generali non risultino per esse appropriati. Anche la definizione di beneficio significativo dovrebbe diventare più stringente. Dovrebbe venire, invece, abolito il criterio basato sui ritorni sugli investimenti, scarsamente utilizzato. La designazione a farmaco orfano dovrebbe avere una durata di sette anni, con la possibilità di estensione a specifiche condizioni in base a richieste giustificate, e potrà essere ritirata su richiesta dello sponsor.